In Vitro Diagnostic Regulation (IVDR)
PentaBase is on track to comply with IVDR
In May 2017, a significant transformation emerged with the publication of the In-Vitro Diagnostic Regulation (IVDR), replacing the EU Commission’s former Directive (IVDD) on in vitro diagnostic medical devices (98/79/EC). Understanding the nuances between IVDD vs IVDR is crucial in navigating the evolving regulatory landscape within the EU.
The arrival of the IVDR signifies more than just regulatory change; it embodies a dynamic commitment to precision, safety, and innovation, shaping the future of in vitro diagnostics. Operating in this space, it’s imperative to stay informed on IVDR regulations to ensure compliance and maintain market access.
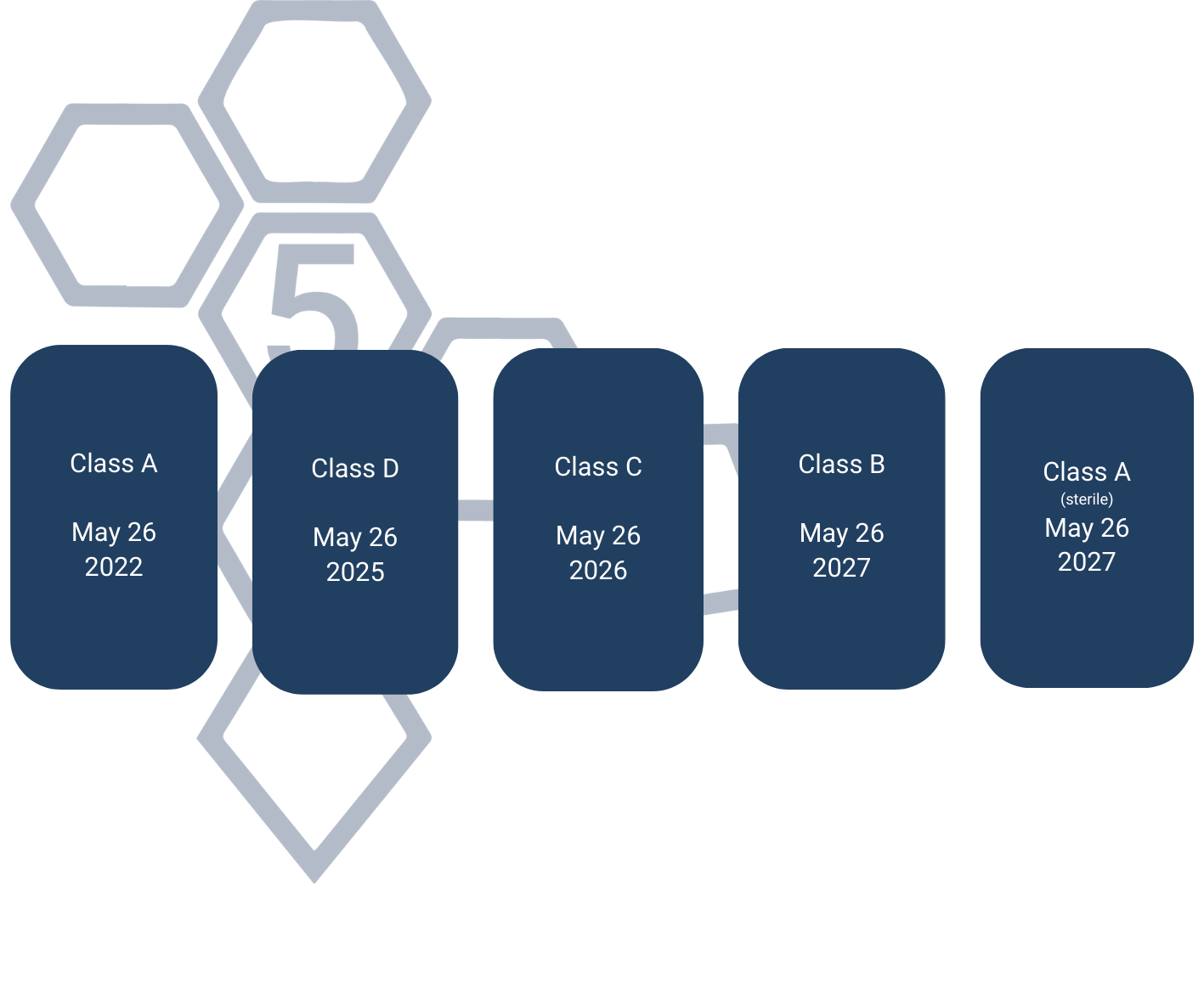
Devices that obtained the CE mark under Directive 98/79/EC retain validity during a transitional period following the IVDR’s implementation, extending until 26 May 2025. General Devices and other products not reviewed by a Notified Body under the IVDD face new requirements under the IVDR. The regulation mandates a phased transition based on each device’s risk classification.
Making the transition from IVDD to IVDR
At PentaBase A/S, we pride ourselves on being at the forefront of delivering cutting-edge in vitro diagnostic solutions that meet and exceed the highest standards of quality and reliability. We are actively recertifying our product portfolio under the new IVDR legislation. This ensures our continued compliance and market competitiveness.
Our commitment to quality management is evident in our QMS adherence to both ISO 13485:2016 and IVDR standards. By meeting these stringent requirements, we uphold strong quality standards. This assures customers of our products’ safety, efficacy, and regulatory compliance.